A set of programs to attempt local methylation-aware realignment around indels. This would be most useful in cases where one is performing some type of BS-seq on a human and wants to decrease incorrect methylation calls due to indel-induced misalignments. These programs work by:
- Finding InDels in alignments and marking their extent. A MAPQ threshold can be applied to alignments before considering whether they support the existence of an InDel.
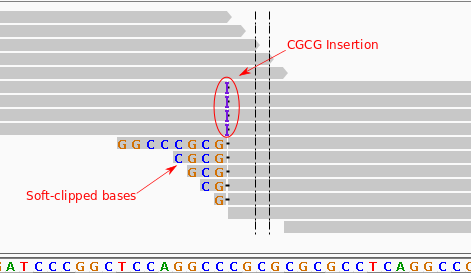
Figure 1 Above is an example of an ROI (region of interest), where there's an insertion of CGCG versus the reference sequence. Note that some alignments are soft-clipped due to partially or totally overlapping the insertion. Since the insertion and the sequence immediately following it are the same, some reads are able to align to the reference without soft-clipping, though their 3'-most alignments are likely incorrect (they're more likely to map to the insertion).
- Tracking the number of alignments supporting a given InDel and using that to filter possible ROIs (regions of interest) where realignment should be performed.
- Looping through the input BAM/CRAM file and finding alignments that overlap an ROI.
- The sequence of both the reference and alignments within a 2*Kmer window centered at the ROI (see for example here, with K=21) are used to create a de Bruijn graph. A graph of the ROI shown above is available in here. The graph starts at the top and proceed through the bottom. Note that there are 6 possible paths through this graph. Blue arrows simply denote paths encountered before during depth first search. For the sake of memory efficiency, the nodes of the graph are stored in a count-min sketch, with 4 bit counters.
- All paths (possibly without cycles, if --noCycles is used) from 2*Kmer before the ROI to 2*Kmer are found and alignments covering the ROI are then aligned to these paths.
- Each alignment from the BAM/CRAM file is assigned to the path to which it best aligns and to which the maxmimum number of other alignments best aligned (in essence, this is an expectation maximization step wherein all alignments covering an ROI are used to weight the final alignment of each other).
- The paths are aligned back to the reference sequence and alignments from the BAM/CRAM file are updated as needed to modify their start positions and CIGAR strings.
{kind=link}
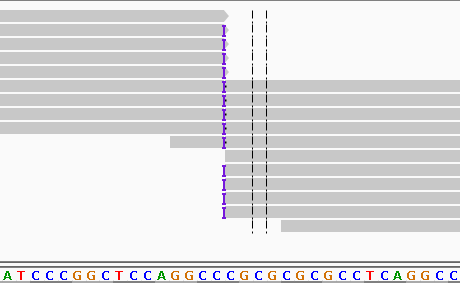
Figure 2 Realigned results are more probable than those prior to alignments. Note that now there are no soft-clipped bases and all of the alignments that can align to the insertion do so.
Note that while efforts are made to keep the output file sorted, there are likely edge cases where the output isn't maintained. You can alternatively pipe to samtools sort, though this will often degrade performance.
MethIndelRealigner is similar to BisSNP's indel realigner function, but runs in a fraction of the time (specifically, the target creator is >10x faster and the realigner is 2-3x faster). The resulting realignments tend to be similar between the two tools.
Paths are aligned to the reference sequence using a global alignment (Needleman-Wunsch) approach. BAM/CRAM alignments are realigned to paths using a global ungapped alignment approach (since there will exist a path to which an ungapped alignment can be made). This is significantly faster than regular Needleman-Wunsch.
Note that murmur3.c and murmur3.h are C implementations of MurmurHash. The C implementation is from Peter Scott and MurmurHash itself is by Austin Appleby. Both of these are in the public domain.
MethIndelRealigner is relatively easy to download and install. The steps are as follows:
git clone https://github.com/dpryan79/MethIndelRealigner.git
cd MethIndelRealigner
git submodule init
git submodule update
make
make install PREFIX=/path/to/desired/installation/location
You can find a simple example dataset under the "example" directory. Note that while MethIndelRealigner TargetCreator doesn't require a sorted BAM file, MethIndelRealigner realign does. The following commands illustrate a typical workflow:
- MethIndelRealigner TargetCreator example.sorted.bam > example.bed
- MethIndelRealigner realign -l example.bed -@ 4 example.sorted.bam example.fa output.bam
- samtools index output.bam
The resulting output.bam file can be directly used for methylation extraction (e.g., by PileOMeth). You can also request that MethIndelRealigner realign index the BAM file by specifying the --index option. Note that the -@ option is overkill for the example, but very useful for real datasets (the slowest part of creating the output file is compression).
Note that in a real dataset, you're likely to see warnings like the following:
[processReads] Skipping 1:631775-631782, too many reads
[realignHeap] Skipping 1:1554209-1554342, too many paths!
The first line indicates that a given region has too many alignments to be processed. This can be altered with the -D option. The default maximum number of alignments a region can have is 1000. In general, it is inadvisable to change this. The reason is that a region with 1000x or more coverage is likely to be dominated by PCR duplicates...creating likely false methylation metrics there. The second warning message indicates that a particular region has too many possible haplotypes. This typically occurs in regions of very low complexity when many of the alignments are of low quality. Again, these regions should likely be ignored during later methylation extraction.
MethIndelRealigner is generally much faster than BisSNP. As a test, bison was used to align a publically available human RRBS dataset (SRR1182519). The resulting BAM files were sorted and read groups added (N.B., MethIndelRealigner does not need these, but BisSNP does). This file was used for target creation with the following commands:
- java -Xmx10g -jar BisSNP-0.82.2.jar -T BisulfiteRealignerTargetCreator -R GRCh38.fa -o SRR1182519.intervals -nt 6 -I SRR1182519.rg.bam
- java -Xmx10g -jar BisSNP-0.82.2.jar -T BisulfiteIndelRealigner -R GRCh38.fa -I SRR1182519.rg.bam -targetIntervals SRR1182519.intervals -o SRR1182519.BisSNP.bam
- MethIndelRealigner TargetCreator -q 1 SRR1182519.bam > SRR1182519.bed
- MethIndelRealigner realign -q 1 -l SRR1182519.bed -@ 4 --quiet SRR1182519.rg.bam GRCh38.fa SRR1182519.realigned.bam
For comparison purposes, samtools view -u SRR1182519.rg.bam | samtools view -@ 4 -bo test.bam - was used to gauge the time taken to simply decompress/compress the BAM file and parse it. The time taken was as follows:
| Command | Time required |
|---|---|
| BisSNP BisulfiteRealignerTargetCreator | 29 minutes 16 seconds |
| MethIndelRealigner TargetCreator | 1 minute 30 seconds |
| BisSNP BisulfiteIndelRealigner | 31 minutes 35 seconds |
| MethIndelRealigner realign | 7 minutes 1 second |
| samtools | 7 minutes |
N.B., BisSNP's target creator would require ~6x more time if run single threaded.
Keep in mind that the tested version of BisSNP seems to ignore the --maxReads option, so its default was used by TargetCreator.
Note that like BisSNP, MethIndelRealigner realign adds OC and OP auxiliary tags to realigned alignments. BisSNP realigned 147,746 and MethIndelRealigner realign realigned 226,478 alignments in this dataset. The two tools disagreed on a number of realignments. MethIndelRealigner did not realign ~63,000 alignments realigned by BisSNP, while BisSNP did not realign ~125,000 alignments realigned by MethIndelRealigner. These ~125,000 differences are due to differences in how paths are aligned back to the reference sequence.
- Using a bunch of spinlocks seems like a wasteful way to multithread. Perhaps we can chaing wake-up between functions with condition variables.
- During graph DFS traversal, only vertices with in-degree >1 need to be tracked. This is similar to a clever memory-saving trick that minia uses. Similarly, switching to a hash would use a little more memory but end up being faster.
- Test memory and speed requirements of just using a hash to store the kmers rather than a bloom filter or count min sketch.
- Use hyperloglog or something similar to estimate the number of unique kmers present in the heap.